About me

I am currently an Associate Professor at HES-SO Valais-Wallis , working mostly on projects for the armasuisse S+T Cyber-Defence Campus (CYD), focusing on the cybersecurity implications of LLMs and cybersecurity technology monitoring. I also teach the introductory class of mathematics and algorithms.
Previosly, I was a post-doc at the Distributed Computing Lab at EPFL and a CYD Distinguished Post-Doctoral Fellow, focused on detecting and countering generative ML use in cyber-offesive operations . Before that, I did my PhD in the Rong Li lab , exploring the molecular mechanisms by which pathogens and cancers can rapidly adapt to stresses (notably drugs), starting with aneuploidy. In turn, that led me to work on developing tools to prioritize potential drug targets to treat breast cancer metastases in Joel Bader and Andrew Ewald labs, as part of the CDT2 consortium , and obtained my MSc and engineering degree from EPFL and Ecole Polytechnique respectively..
More generally, I am interested algorithms for generation, evaluation, and utilization of knowledge, be it in context where the knowledge is derived automatically (such as machine learning models), or is interpreted automatically (such as in ontologies). I am in particular interested by generative, distributed, explainable and secure ML.
X-refs
- Blog (occasional technical, UX and scicom tibits)
- Twitter (inactive post archive)
- Mastodon (infosec readonly alt)
- GitHub
- Google Scholar (Sematic Scholar )
- StackOverflow

Teaching Stochastic Parrots hide-and-seek through normative fine-tuning
Why: While relatively small, the detection of generative machine learning models has been a subject of some research since the release of the GPT-2 language model. However, most for that research has assumed the best-case scenario, with an attacker not attempting any evasion or only a single type. However, an attacker is under no such constraints. In this paper, we explore how a low-budget attacker, armed only with a small dataset and a GPT-2 based generator could attempt to evade on of the state-of-the art generative models detectors - a fine-tuned BERT model.
What: Along with Henrique Da Silva - an EPFL bachelor student - we show that a slight modification to the optimizer used to fine-tune the GPT-2 model and access to the "reference" human speech dataset used to pre-train the BERT model, we are able to fine-tune GPT-2 on less than 10 000 utterances so that it becomes not only effectively invisible to BERT model, but BERT model training fails even if it is detected.
More: TBA
Combining Transformers with Text GANs (Supervisor)
Why: Detecting generative machine learning models in the wild is a two-edged sword. On one hand, we need it to distinguish real human beings from generative models. On the other hand, creating a good detector can in some cases enable the attacker training the generative model to improve it further. It is even the principle itself of Generative Adversarial Networks (GANs). While the best performing generative language models are based on the Transformer architecture, there are a number of them still based on the previous generation of the latent space representation - LSTM (long-short term memory).
What: Along with Kevin Blin - an EPFL bachelor student - we have empirically showed that the transition to the Transformer architecture in GAN setting is not trivial. Even if a quality, transformer-based detector was made public, it could not be directly used to train the generative model to evade it, at least not within the standard text-generating GAN architectures.
More:
- Paper presented and published in RANLP 2021
- Twitter thread trying to distill in a more accessible manner
- An EPFL news article going over the bulk of our findings.
Iterating on loupes: unsupervised approaches to dynamically adjust visual similarity metrics
Why: While image classification with regards to pre-determined classes works great, it scales poorly. Not only human labor to annotate reference image datasets are needed, but past annotations are likely to be rendered irrelevant due to the change of concepts used to define the image classes.
What: In this paper we explore a way to overcome that problem by switching away from classes to clusters in a custom similarity metric space. We then investigate means to adjust that similarity metric to dynamically adjust clusters as new classes emerge or become relevant.
More: TBA
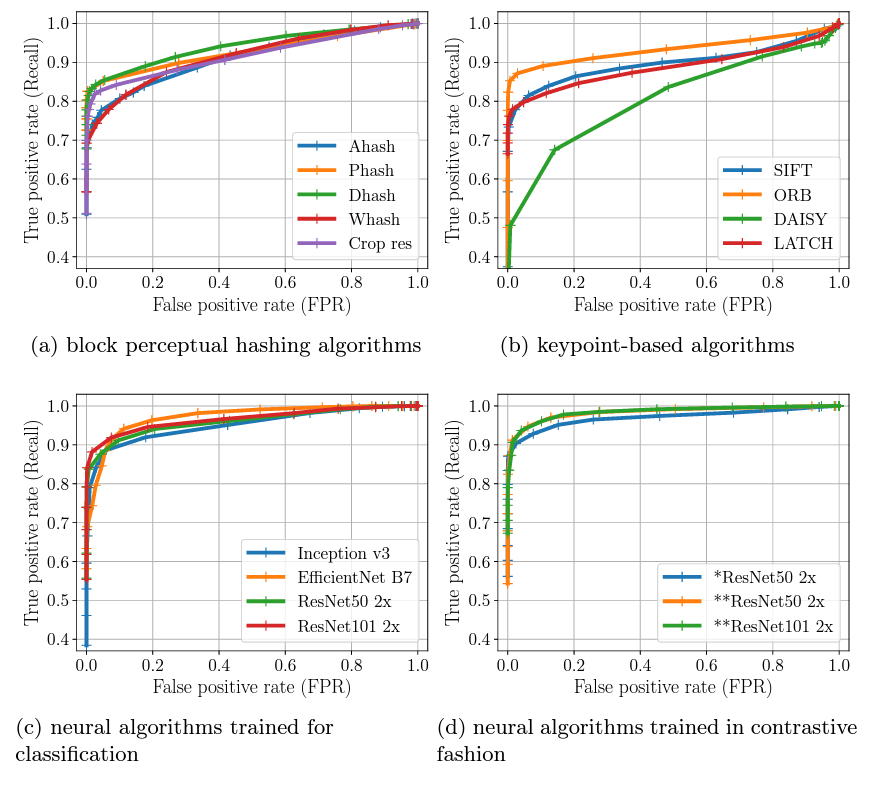
Finding a visual needle in a haystack of images, fast
Why: Researchers who are trying to understand the way visual media is shared and consumed on the internet are faced with a massive problem. They need to process massive amounts of images to extract visual elements that picked up by humans and can potentially be relevant to other social processes, ranging from suicidal ideation to targeted organized harassment. However, the researchers are limited by the computational power at their disposal in a way larger entities that have approached that problem in the past aren't.
What: Along with a CYD intern and an EPFL master student - Cyril Vallez - we developed a benchmark to evaluate the robustness of perceptual similarity metrics. After analyzing the accuracy and resource efficiency of three main families of perceptual similarity evaluation algorithms (perceptual hashing, keypoints, neural hashing), we validated our results on in-the wild images, namely memes shared on Reddit. We found that two relatively little-known methods - perceptual hashing Dhash and neural hashing SimCLR v2 - performed great while keeping resource utilization low. Notably, dHash was not only the most computationally efficient, but outside large image rotations or tearing was one of the most if not the most resilient methods in identifying images.
More:
- Preprint on Arxiv
- GitHub repo containing the benchmark and the benchmarked methods
Efficient distribution of machine learning over non-differentiable manifolds
Why: Deep Machine Learning (ML) have achieved tremendous results in recent years. A lot of them were due to the unreasonable effectiveness of the stochastic gradient descent (SGD) on the problems that could be reduced to differentiable manifolds - such as for instance word embeddings for text generation. However, a number of problems - such as self-driving or strategy games - require discrete actions that cannot be differentiated. Until now, reinforcement learning has proven to be quite successful, but is still challenging to scale and does not deal well with some classes of problems.
What: I proposed a new way of trivially distributing the learning in an efficient and byzantine-resilient manner in case many nodes are able to sample possible solutions efficiently.
More: TBA
Equivalence between SGD and a class of evolutionary algorithms
Why: A lot of major achievements of the Deep ML has been achieved thanks to the unreasonable effectiveness of SGD. Due to that, there has been a lot of research invested into understanding when and why SGD converges and finds good results rapidly. There are approaches that allow to learn effectively on non-differentiable manifolds, such as reinforcement learning, but it is unclear if they have properties as nice as the SGD.
What:I put forwards a proof that showed and equivalence between a class of evolutionary algorithms (GO-EA) and SGD. While less computationally efficient, the proposed EA is much easier to parallelize and do not require an explicitly differentiable manifold - just one that is smooth enough.
More: TBA
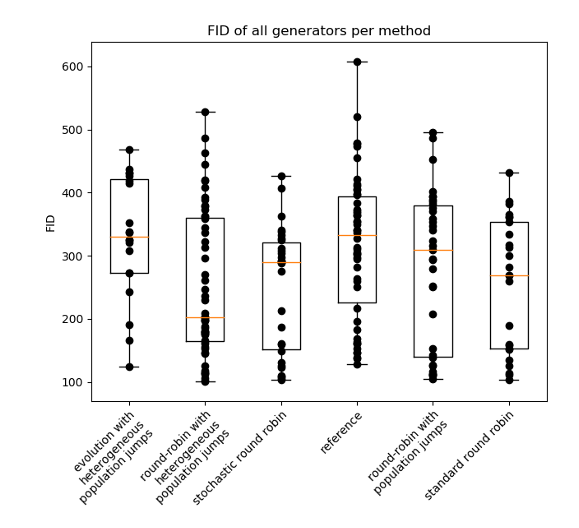
Improving GAN training with population genetics
Why: Since their introduction by Goodfellow in 2014, they have managed to achieve some impressive results in image manipulation. Several alternative architectures aiming to improve the original architecture have been proposed and published. Unfortunately, recent results suggesting that the improved results resulted from selection of best samples and numerous restarts of the training process.
What: We managed to show that some practices inspired by the theory of evolution and co-evolution allowed to stabilize the training of GANs and achieve better results with lower computational expenses.
More: Preprint on Arxiv
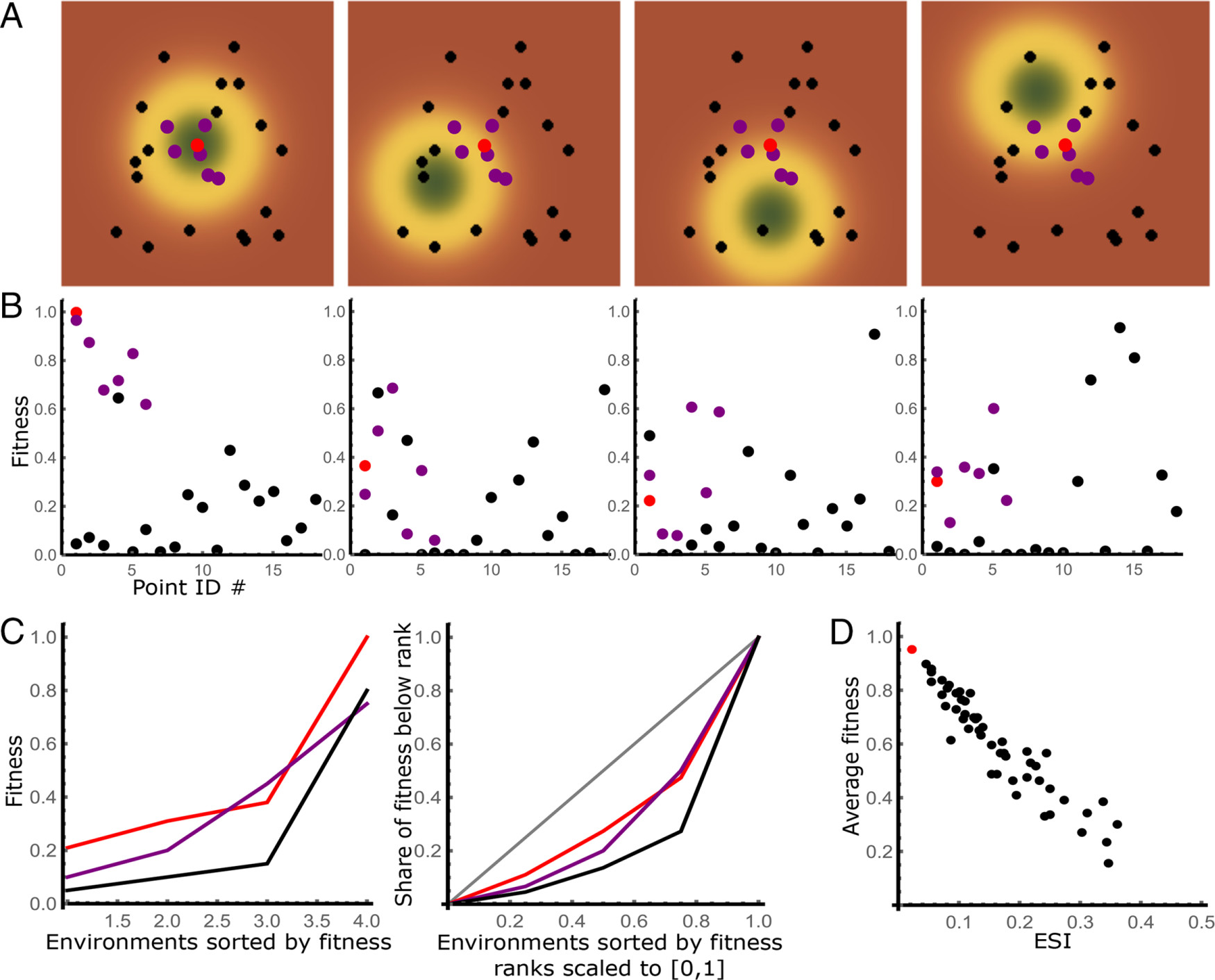
Detecting generalists in a heterogeneous population
Why: One of the main problems with treating cancers are the small population of cancer cells that survive treatments that kill most of the tumor, and re-start the tumor growth following a treatment - sometimes years down the line. A common explanation are so-called "cancer stem cells" - cells that reproduce slowly and inhabit well-protected niches.
What: Performing a re-analysis of a growth assay in stress conditions of the breast cancer cell lines (aka in presence of drugs), we have noticed that the pattern was actually different. Some cells performed great in some conditions, terribly in others - as if they were covering different regions in a latent space where drugs were acting in. The few cell lines that seemed to perform well across the board didn't have the drug resistance features that would be expected in the stem or stem-like cells. Combined with prior findings in yeast, I was able to formalize a model of adaptive evolution related the the Fisher Geometrical model, leading to a tool that's able to estimate the dimension of the latent space, combinatorial therapies that we would expect to be most efficient, as well as point towards the generalists - strains most likely to survive after combinatorial therapies.
More:
- Paper presenting the findings
- More experimentally oriented paper
- My PhD thesis on the topic, going over the bulk of our findings.
Most likely amino acids in the Last Universal Common Ancestor
Why: Today, the life is organized into three main branches - Eucaryotes, Bacteria and Archeobacteria. However, the similarity in genetic code across the three main branches, strongly points to a time when they were one - probably a single organism - commonly referred to as the Last Universal Common Ancestor (LUCA). What can we say about the genome of this organism?
What: Building on top of the past research into the minimal likely genome that is common to all living organisms on earth, we have discovered that it is likely that LUCA did not have two of the amino acids all the living organisms share today. Which poses the question - did they emerge as a result of a convergent evolution? Emerge shortly before the three main branches split off? My role in this project was to write software that was performing automated homology analysis.
Paper presenting the findings(Biological) business logic-first image analysis
Why: Despite numerous tools to perform numerical analysis of microscopy images, they tend to rely on programming competences and understanding of underlying numerical methods of end users. Which is an obstacle in their wide-spread adoption and correct application by the experimental biology community.
What: I have distilled a natural language of queries my biological colleagues had when it came to analyzing the microscopy images for their projects throughout my PhD. The resulting language is built on top of Python and operates using natural primitives, as well as providing straight-jackets to make sure that no accidental p-hacking can occur. It's modular and modules can be added by collaborators specializing in numerical methods.
More: a GitHub repository of the source code with examples of several applications.
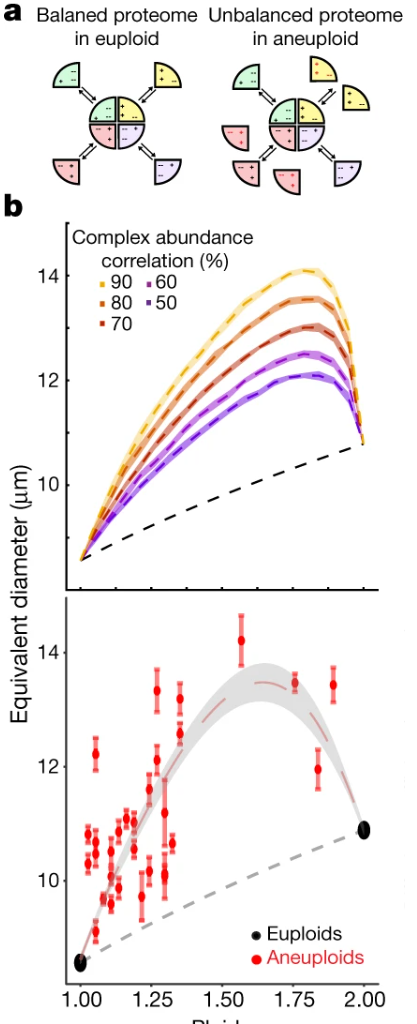
Osmotic pressure in aneuploid yeast
Why: Aneuploidy is a common feature of cancer cells and - in many cases - correlate with adaptive stress response in other organisms. However, there is a debate whether aneuploidy is adaptive or nefarious for the cells. In the later case, it could be exploited to kill aneuploid cancer cells more efficiently and create drugs for cancer that would promote aneuploidy. However, that would require the understanding of mechanisms by which aneuploidy stresses out the cells of the organism
What: My colleagues were able to demonstrate that aneuploidy-induced stress correlates with osmotic shock response in baker's yeast, and further demonstrate that aneuploid cells were in fact in a hyper-osmotic state. However, it was not clear why. My contribution, was to use foundational thermodynamics to demonstrate that such an over-pressure was expected in case of any aneuploidy - through transcriptional imbalance and protein functioning mainly through complexes, and provide simulations that quantitatively reproduced the experimental results.
More:
- Paper presenting the findings
- Arxiv preprint discussing more in depth divergence with prior results, that suggested aneuploidy-induced stress was instead due to translation machinery overload .

Random assembly of the cytoskeleton in yeast
Why: My colleagues have experimentally observed that the size of the actomyosin ring that is required for the budding yeast division scaled in contraction speed with the size of that ring - itself correlated with the size of the mother cell. the size of the cell, which was not easy to explain with then dominant hypothesis of subunits assembly. Which was even less clear, is how the ring count contract upon removal of contractile motors - myosin.
What: My contribution was to propose that actomyosin strains were assembled randomly and that contraction was driven not only by the myosin motors, but also through the depolymerization of antisense actin strands in the presence of cross-likers able to uniformly diffuse along the lengths of the strands.
Paper presenting the findingsOptimal hypothesis formulation based on network representation of prior knowledge
Why: Despite a number of ad-hock suggestions on how network theory could be used to elucidate biological mechanisms, there is still no understanding why they should work at all, or which versions of network biology algorithms work well.
What: I have showed that a version of network biology algorithm is equivalent to the formulation of most likely and most parsimonious molecular mechanism underlying a phenotype of interest, with regards to the existing biological knowledge.
More: a GitHub repository providing implementation of the algorithm in the context of biology
Biological systems as universal approximators
Why: Despite extensive investigations into how the biological organisms operate, there has been little research as to why they operate this way - or, as a matter of fact, have emerged at all in the chaos of primordial chemical reactions.
What: We approach biological organisms as universal approximators - a term that has been coined in the process of trying to better understand deep neural networks. We show that this approach not only makes sense, but also explain a number of wierednesses in biological organisms and allows us to fairly accurately predict the number of essential genes in yeast, abstracting away their exact biological function and treating them as just "knobs" that can be turned to best adapt to the environment.
More: Prepring on BioRxiv
Mitochondrial pathways of misfolded protein degradation
Why: My colleagues in the Rong Li lab have made an observation that misfolded proteins in heat-shocked bakers yeast would tend to accumulate on the mitochondria. Why?
What:We have eliminated the hypothesis it could have been due to the electrostatic thermodynamics reasons and linked it to the RNA-rich stress granules. Upon further investigation, my colleagues have discovered that they were formed due to the import of misfolded proteins by mitochondria, where they were degraded. This mechanism also exists in mammalian cells, potentially explaining how mitochondrial defects tend to correlate with neurodegenerative disorders due to misfolded protein accumulation, such as Alzheimer.
More: Paper summarizing biological findings
Network systems biology for polypharmacology
Why: Most of the drugs in development and on the market today do not bind a single target, but rather have an affinity for a number of targets. While this is not a great news for a single pathway - single disease - single target - single molecule paradigm, and in some cases result in secondary effects, it also means that a lot of drugs can be repurposed for neglected diseases. However, we need to figure out which ones can potentially be and prioritize them.
What: I was able to do a proof of concept that used a systems network biology representation of existing biological knowledge to use the results of in-silico binding assays to infer potential off-target activity.
Outside research, I sometimes play with data to answer a common question over on Quora, such as for instance about PISA rankings and what it means for France, or do scientific outreach in domains I am familiar with, such as 2014 Ebola outbreak COVID, origins of COVID , vaccines (also here), HCQ for COVID (also here), chemical thermodynamics or Simpson's paradox.

In my free time, I run, swim and bike in summer and ski in winter, or try to teach my cats tricks. So far they are doing a better job at teaching me to do tricks for them instead.
You can contact me at:
Online, you are welcome to send me a message over on LinkedIn or Twitter or to send a mail through the EPFL directory.